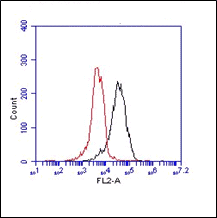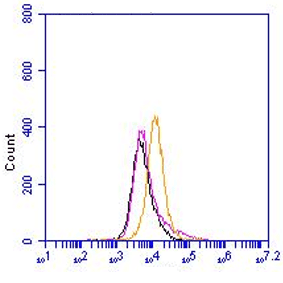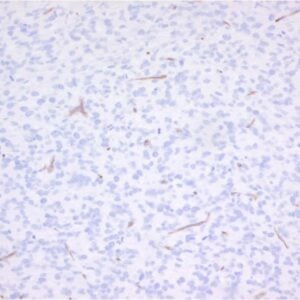




The monoclonal antibody 1.5D7 recognizes the disease associated isoform of the prion protein
termed PrPSc. Prion diseases, also known as spongiform encephalopathies, are a group of
neurodegenerative diseases that include BSE (bovine spongiform encephalopathy) in cattle, scrapie
in sheep and CJD (Creutzfeldt-Jakob disease) in humans. The normal cellular form of the prion
protein is denoted as PrPC and is a constitutively expressed glycosylphosphatidylinositol anchored
protein that has been shown to play a role in myelin formation. PrPC has an unstructured N-terminal
part and a C-terminal part consisting of three α -helices and two short β strands. Refolding of the
normal prion protein results in PrPSc, which has a tightly packed C-terminal part enriched in beta
sheets which is insoluble and resistant to digestion by proteases. Prion diseases are characterized by
the deposition of highly structured aggregates of PrPSc, astrocytosis, neuronal cell death and
spongiform structures in the brain. These diseases can be sporadically (unknown cause), be inherited
due to polymorphisms or mutations in the prion protein gene or be transmitted by an infectious particle
which is believed to consist of PrPSc only. In order to study prion diseases the detection of PrPSc and
the ability to discriminate between the normal and disease associated PrP is of pivotal importance.
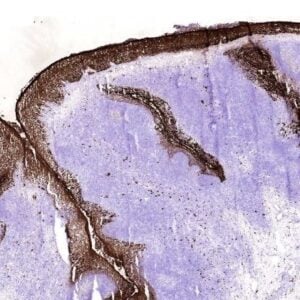
The monoclonal antibody 1.5D7 can be used for the specific identification and characterization of
PrPSc in tissue sections by immunohistochemistry and PET-blot.
IHC-P: Paraffin sections were deparaffinized, rehydrated and endogenous peroxidase was quenched using 0.3% H2O2 in methanol for 20 min. As positive control BSE infected brain tissue was used and as negative control non-diseased brain tissue (Ref.1).

 Powered by Bioz
Powered by BiozRelated products
-
View product €139.00 €333.00Price range: €139.00 through €333.00
-
View product €139.00 €9,220.00Price range: €139.00 through €9,220.00
-
View product €530.00
-
View product €139.00 €431.00Price range: €139.00 through €431.00
-
View product €139.00 €431.00Price range: €139.00 through €431.00
-
View product €374.00
-

ICAM-1, Human, pAb, biotinylated
Cross reactivityBaboon – Yes, Chimpanzee – Yes, Cynomolgus monkey – Yes, Rhesus monkey – YesView product €274.00 -
View product €139.00 €474.00Price range: €139.00 through €474.00
-
View product €139.00 €333.00Price range: €139.00 through €333.00