The Complement System
Every day, our immune system protects us from harmful microbes and infections. Most of these challenges remain undetected due to the continuous protection provided by the complement system.
What is the Complement system?
The complement system is an essential cascade of over 30 proteins that triggers powerful immune responses. When activated, it produces:
- Anaphylatoxins (C3a, C5a) that orchestrate responses from chemoattraction to apoptosis
- Membrane Attack Complex (MAC/C5b-9/TCC) that creates pores in pathogen membranes, causing cell death
Originally recognized for innate immunity, complement also activates adaptive immunity (T and B cells), maintains immunological memory, and plays roles in tissue regeneration, tumor growth, autoimmune disorders, kidney disease, neurodegeneration, and cancer.
Quick links
Related products
Explore our products today and take the next step in utilizing the power of complement science for breakthrough medical advancements.
TCC, Human, ELISA kit
C5, Mouse, mAb BB5.1
3 Complement Pathways
The Classical Pathway: Antibody-Initiated Defense
Initiated when C1q binds to antigen-associated IgM or IgG hexamers, forming immune complexes. This represents your adaptive immune system recruiting complement as a powerful amplification mechanism: antibodies mark the target, and complement destroys it.
The Lectin Pathway: Pattern Recognition
Activated upon recognition by specific molecules, including mannose-binding lectin (MBL), collectins, and ficolins). These pattern-recognition molecules bind to microorganism-associated molecular patterns (MAMPs) or carbohydrate structures on damaged cells, enabling immediate response without requiring prior antibody production.
The Alternative Pathway: Constant Surveillance
Undergoes continuous low-level activation through spontaneous C3 hydrolysis (“tick-over”), enabling rapid response on surfaces lacking proper regulation, typically non-host structures. Serves as both independent activation and powerful amplification loop for the other pathways.
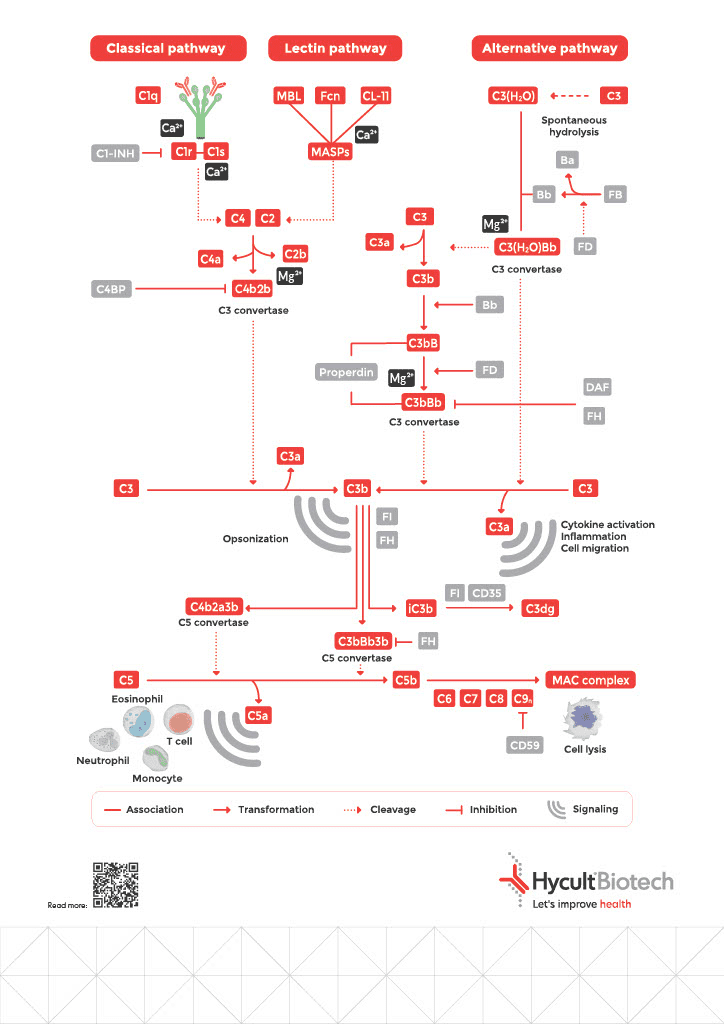
The Critical Balance: Regulation Prevents Self-Destruction
Complement is powerful and constantly active. Unchecked, it would destroy your own tissues as efficiently as pathogens.
To prevent complement overactivation, the system is tightly regulated by soluble and membrane-bound proteins that serve as checkpoints limiting excessive activity. For each type of activated fragment, there is at least one inhibitor or inhibitory mechanism. These regulators act by accelerating breakdown of C3 and C5 convertases or by preventing MAC formation. Most regulators are encoded by genes located in the RCA (Regulators of Complement Activation) gene cluster.
When Regulation Fails: Complement Dysregulation and Disease
Insufficient regulation can lead to excessive activation, resulting in serious conditions such as thrombo-inflammation, immune dysregulation, vascular endothelial inflammation, and organ damage.
Genetic or acquired deficiencies in complement inhibitors or their cofactors can disrupt balance of complement regulation, leading to uncontrolled activation characterized by persistent opsonization, MAC-mediated cell lysis, and chronic inflammation.
Such dysregulation is associated with a range of immune-mediated diseases affecting the eyes, kidneys, blood, and nervous system.
Emerging Clinical Applications and Associated Challenges
Recent clinical milestones, such as the approval of eculizumab for generalized myasthenia gravis, underscore the system’s therapeutic versatility beyond traditional indications like atypical hemolytic uremic syndrome (aHUS). Complement modulation is now being explored in complex disorders:
- Neurodegeneration (Alzheimer’s, ALS, Parkinson’s)
- Autoimmune disorders (lupus, rheumatoid arthritis)
- Ischemia-reperfusion injury
- Transplant rejection
- COVID-19-associated thromboinflammation
- Cancer immunotherapy
The challenge? Achieving localized modulation without systemic immunosuppression.
The Three Critical Factors for Reliable Complement Measurement

Want to Measure Complement Accurately?
Since 1994, Hycult Biotech has addressed measurement challenges with assays and antibodies delivering reliable, reproducible results:
- Low batch-to-batch variability through strict manufacturing controls
- Validated specificity with published cross-reactivity data
- Standardized protocols optimized through complement-specific expertise
- Neo-epitope recognition targeting sites exposed only after activation
Whether in research or clinical settings, our tools ensure your measurements reflect biology, not artifacts.
Frequently asked questions
Yes, we can provide you with a poster summarizing the entire pathway. For further reading regarding complement function, regulation and its role in disease, we recommend the following review papers:
Brandwijk Ricardo J. M. G. E. , Michels Marloes A. H. M. , van Rossum Mara , de Nooijer Aline H. , Nilsson Per H. , de Bruin Wieke C. C. , Toonen Erik J. M. ,Pitfalls in complement analysis: A systematic literature review of assessing complement activation, Frontiers in Immunology, Volume 13 – 2022, 2022, 10.3389/fimmu.2022.1007102
Mastellos, D.C., Ricklin, D. & Lambris, J.D. Clinical promise of next-generation complement therapeutics. Nat Rev Drug Discov 18, 707–729 (2019). https://doi.org/10.1038/s41573-019-0031-6
John P. Atkinson, Terry W. Du Clos, Carolyn Mold, Hrishikesh Kulkarni, Dennis Hourcade, Xiaobo Wu, 21 – The Human Complement System: Basic Concepts and Clinical Relevance, Editor(s): Robert R. Rich, Thomas A. Fleisher, William T. Shearer, Harry W. Schroeder, Anthony J. Frew, Cornelia M. Weyand, Clinical Immunology (Fifth Edition), Elsevier, 2019, Pages 299-317.e1, ISBN 9780702068966
Intracellular complement functions as an autonomous regulatory layer that modulates immune cell metabolism, survival, and inflammatory responses, operating inside lysosomes and mitochondria. In contrast, extracellular complement primarily functions in immune surveillance and pathogen elimination via the three pathways in plasma and tissues. A few examples in which intracellular complement differs from extracellular [3]:
| Extracellular | Intracellular |
| Activated in plasma/tissue fluids by classical, lectin, or alternative pathway | Activated within lysosomes, cytosol, or mitochondria |
| Produces C3a/C5a and MAC for immune defence | Produces C3a/C5a to regulate metabolism and inflammation |
| Requires convertase formation for C3/C5 cleavage | May bypass convertases, by using local proteases |
Developing complement inhibitors for clinical use is challenging due to complex pathway crosstalk and activation bypass mechanisms. Non-canonical proteases such as thrombin or kallikrein can cleave C3 or C5 without convertases, allowing bypass activation. This can undermine the efficacy of convertase-targeting therapies [3]. Moreover, functional overlap between pathways enables activation even when one route is blocked. Another challenge lies in the genetic variation in regulatory proteins like factor H and CD46, which can alter disease susceptibility and patient response to therapy. Pathogens further complicate treatment by mimicking host complement regulators, enabling immune evasion. Intracellular complement activity adds complexity and raises questions about how complement inhibitors are expected to work in clinical practice.
Complement dysregulation plays a central role in the development of aHUS and C3 glomerulopathy. In these diseases, uncontrolled alternative pathway activation leads to excessive C3 fragment deposition on host tissue. This results from mutations or autoantibodies affecting regulators such as factor H, factor I, CD46, or factor H-related proteins. In aHUS, factor H variants often cluster in the C-terminal domains responsible for host-surface recognition. This impairs regulation on endothelial cells, promoting thrombotic microangiopathy. In contrast, C3 glomerulopathy-associated variants localize to N-terminal domains affecting fluid-phase C3 regulation. The imbalance causes persistent C3b deposition and complement amplification in the glomerular basement membrane. (The glomerular basement membrane is an extracellular matrix in the kidney.) This drives inflammation, tissue injury, and renal dysfunction. Both diseases reflect distinct but overlapping signatures of alternative pathway dysregulation. Genetic insights from patients have clarified how regulatory defects trigger complement-mediated kidney pathology. [3]
Numerous diseases have been associated with complement system dysfunction. Below is a selection of some widely discussed examples:
- Kidney-Related Diseases (C3G, Lupus Nephritis, aHUS, MN): Characterized by complement dysregulation, immune complex formation, and inflammation, leading to glomerular damage, renal dysfunction, and progressive kidney disease.
- Hematological Disorders (PNH, AIHA, TMA): Conditions involving red blood cell destruction, thrombotic complications, and hemolytic anemia due to impaired complement regulation and immune system dysfunction.
- Neurological Diseases (Multiple Sclerosis, Alzheimer’s Disease, NMOSD, MMN, ALS, Parkinson’s Disease): Chronic neuroinflammatory and neurodegenerative disorders where complement activation contributes to neuronal damage, demyelination, and disease progression.
- Sepsis, SIRS & Ischemia-Reperfusion Injury (IRI): Dysregulated immune activation leading to systemic inflammation, organ dysfunction, and tissue ischemia, relevant in conditions such as stroke, myocardial infarction, and trauma.
- Systemic Autoimmune Disorders (SLE, TMA-related conditions): Autoantibody formation and complement activation drive widespread inflammation, immune complex deposition, and increased disease severity.
- Oncology & Tumor Progression: Complement activation, particularly TCC formation, plays a role in the tumor microenvironment, influencing immune evasion, chronic inflammation, and cancer progression. Complement inhibitors are being explored as potential therapeutic strategies in oncology.
Because of the large number of complement-mediated diseases and recent advancements in large-scale genomics and proteomic research, interest in the complement system has been revitalized, especially as a promising target for therapeutic intervention. This was demonstrated more than a decade ago by eculizumab (Soliris, Alxion), the first complement-specific drug designed to inhibit the key complement component C5.
Therapeutic strategies can target different levels of the complement pathway to prevent or control excessive activation. A central approach is the use of monoclonal antibodies against C5, such as eculizumab, to prevent C5a and membrane attack complex formation. Compstatin analogues act upstream by inhibiting C3 activation, blocking both opsonization and downstream amplification. To target the alternative pathway specifically, inhibitors of factor B or factor D disrupt its amplification loop. Some therapies interfere with convertase assembly or stability, thereby reducing the generation of effector molecules. C5a receptor (C5aR1) antagonists selectively block inflammatory signaling without affecting upstream complement functions. Complement regulation can also be restored by engineered regulators that mimic the activity of proteins like factor H or CD46. These specific targeted deliveries improve a way to modulate complement activity.
The complement system plays an important role in therapeutic research because of its contribution in immune defense, inflammation, and disease pathology. Malfunction of complement activity contributes to autoimmune diseases, inflammatory disorders, neurodegeneration, cancer, and infectious diseases, making it an interesting target for drug development.
Contact us for more information
At Hycult Biotech, we recognize the growing demand for advanced complement research tools that facilitate accurate analysis and targeted intervention in immune-related diseases. Our portfolio includes high-quality complement pathway inhibitors (how to inactivate complement), complement assay kits, and monoclonal antibodies designed to support drug discovery and translational research. On our website, you can search for products that match your research needs. Additionally, we are happy to provide expert advice. Feel free to contact us via the contact form.