Understanding the Alternative Pathway in Complement Activation
The complement system is an essential component of the innate immune system, providing a first line of defense against invading pathogens. It consists of approximately 50 plasma and membrane-bound proteins that interact in a tightly regulated network of activation, amplification, and regulation. Once activated, complement promotes pathogen elimination through opsonization, recruitment of inflammatory cells, and direct lysis via the membrane attack complex. Complement activation can be initiated through three activation routes: the classical (CP), lectin (LP), and alternative pathway (AP). While the CP and LP are triggered by recognition of immune complexes or specific carbohydrate patterns, respectively, the AP has a unique role as a continuously active surveillance mechanism. The AP is maintained in a state of low-level activation through the spontaneous hydrolysis of C3, a process often referred to as “tick-over.” This allows the AP to serve as an immediate defense-mechanism once a pathogen surface is encountered, ensuring a swift immune response. In addition to protecting against pathogens the AP also plays a crucial role in clearing apoptotic cells, preventing biomaterial-induced inflammation, limiting tissue damage, and participating in cancer immune surveillance. Given the broad involvement of the AP and its spontaneous activation in both physiological and pathological processes, it is of utmost importance to unravel the mechanisms underlying its activation and regulation in health and disease.
This page provides an overview of the regulatory mechanisms controlling AP activity, the consequences of its dysregulation, and the challenges associated with accurately measuring AP function. Furthermore, we provide an overview of known AP-related diseases, offering insights into its clinical relevance and diagnostic value.
We are glad to support you!
Take advantage of our dedicated support team for any technical assistance you need while using our products or considering them for your research needs.
- Original supplier of innate immunity products since 1994.
- Our commitment to quality: Read more!
- Contact your local distributor for ordering.
- Contact our support team for more information.
Activation and Regulation Mechanisms of the Alternative Pathway
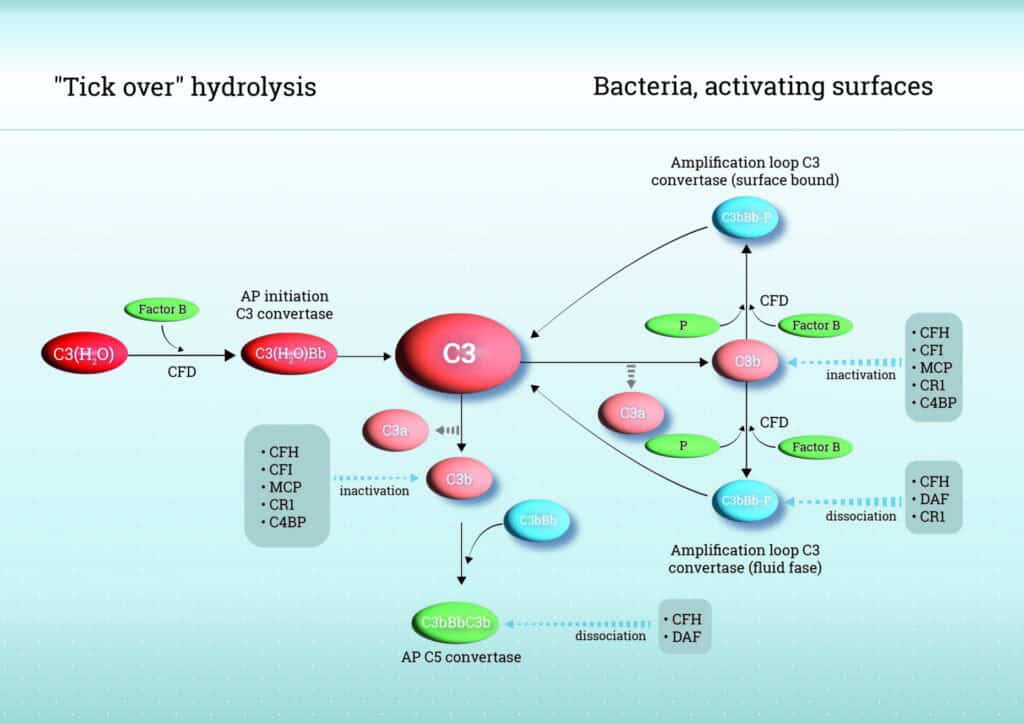
As mentioned above, unlike the classical and lectin pathways, AP does not rely on specific recognition molecules but is continuously active through a process known as “tick-over”. This activation is initiated by the spontaneous hydrolysis of the internal thioester bond in complement component C3, producing C3(H2O) in the fluid phase. C3(H2O)binds to Factor B (FB), which is then cleaved by Factor D (FD), forming the fluid-phase C3 convertase (C3(H2O)Bb). This enzyme cleaves additional C3 into C3b and C3a, amplifying the response. When C3b covalently attaches to nearby surfaces, such as microbial membranes, it initiates surface-bound amplification by additional binding of FB, forming C3bBb complexes, thereby amplifying the response. Properdin (Factor P), the only known positive regulator of complement, binds to and stabilizes the C3bBb complexes, significantly prolonging its half-life and thereby enhancing robust complement activation and opsonization. Due to its continuously active state, the AP requires strict regulation to prevent overactivation which can lead to excessive inflammation, tissue damage, and complement-mediated disease.
Control of AP activity is achieved by a network of soluble and membrane-bound regulators. Factor H (FH) is a central soluble regulator, competing with factor B for C3b binding, accelerating decay of the C3 convertase (C3bBb), and acting as a cofactor for Factor I (FI)–mediated cleavage of C3b into inactive fragments. FHL-1 (FH like-1), a smaller splice variant of FH, shares these regulatory functions and provides tissue-specific protection, while Factor H-related proteins (FHR-1, FHR-2, FHR-3, FHR-4, and FHR-5) can modulate AP activity by competing with FH and FHL-1 for C3b binding, fine-tuning complement regulation, and, in certain contexts, contributing to dysregulation and disease.
Next to FH and its family members, membrane regulators including complement receptor 1 (CR1/CD35), membrane cofactor protein (MCP/CD46), and decay-accelerating factor (DAF/CD55) protect host cells: CR1 combines decay-accelerating and cofactor activities, MCP supports FI-mediated C3b inactivation, and DAF accelerates convertase decay. This strict balance between activation and regulation ensures efficient immune defense while minimizing host tissue damage. See Figure 1 for a schematic overview of the AP.
Consequences of Failed Alternative Pathway Regulation
Dysregulation of the AP leads to several complement-mediated diseases, highlighting its delicate balance between host protection and pathological inflammation. Atypical hemolytic uremic syndrome (aHUS) results from complement-mediated endothelial damage, causing thrombotic microangiopathy and acute kidney injury. C3 glomerulopathy (C3G), including dense deposit disease (DDD), is driven by excessive C3 deposition in the glomeruli, leading to progressive kidney injury and potentially end-stage renal disease. Also lupus nephritis (a severe manifestation of systemic lupus erythematosus (SLE)) is increasingly recognized to have a pathogenic role for alternative pathway activation [1][2].
In addition to kidney diseases, AP dysregulation contributes to other complement-mediated disorders. Age-related macular degeneration (AMD) is a leading cause of vision loss in the elderly and is associated with excessive local complement activation in the retina. Dysregulated AP activity contributes to chronic inflammation, accumulation of drusen deposits, and damage to retinal pigment epithelial cells, ultimately leading to progressive retinal degeneration and vision impairment.
Impaired function or regulation of Factor H (FH) is a key driver of these disorders. Dysregulation can arise from genetic mutations, polymorphisms, competition by Factor H–related proteins (FHRs), or the presence of anti-FH autoantibodies, all of which reduce the ability to inactivate C3b and control C3 convertase activity. The resulting uncontrolled alternative pathway activation leads to local tissue inflammation, deposition of complement fragments, and progressive damage in organs such as the kidney or retina. Next to these established AP-mediated diseases, recent studies also suggest that dysregulation of Factor H (FH) may have broader systemic consequences. Anti-FH autoantibodies have been identified in a range of autoimmune conditions, indicating that FH disruption can drive complement-mediated pathology beyond the traditionally recognized disorders [3]. These findings underscore the central role of FH and its family members in maintaining complement homeostasis and highlight them as important targets for emerging therapeutic strategies.
For a more comprehensive list of alternative pathway-related diseases, please check the FAQ section. These diseases illustrate how delicate the balance is between protective immunity and pathological inflammation.
Clinical relevance and the need for reliable measurements
The growing recognition of the AP in a wide range of pathological processes has generated increasing interest in accurately measuring its activation. Clinical advances, such as C5 inhibitor eculizumab and the development of upstream AP-targeted drugs like FB and FD inhibitors in complement-targeted therapies underscore the importance of reliable detection of AP-specific markers, like Factor B, D and H, both for diagnostic and therapeutic monitoring purposes.
Given its pivotal role, FH is being explored both as a therapeutic target and a biomarker for disease. Measuring FH and FHR levels in circulation is emerging as a powerful approach to improve diagnostics, predict disease progression, and monitor complement-targeted therapies.
Despite progress in measuring AP markers, several challenges remain. Activation of the complement pathway is highly sensitive to artificial activation during or after sample collection, thereby obscuring the results. In clinical practice, this sensitivity can be both beneficial and a challenge. Complement reacts quickly to infections, tissue injury, or immune dysregulation, making it a valuable tool for early diagnosis. Its rapid changes during disease progression or recovery also enable physicians to closely monitor treatment effects.
As mentioned, an increasing number of diseases are being recognized in which the AP appears to play a role. However, its exact contribution often remains unclear, highlighting the need for further research. Key questions include: Is AP overactivation the cause of the disease, or does the disease itself trigger AP overactivation? And could the AP serve as a therapeutic target in these conditions?
To address such questions, it is essential to measure AP activity at the level of individual markers. Hycult Biotech offers a wide range of tools to assess AP markers, including specific pathway components, activation markers, and regulators. Please refer to our related products to identify the most suitable marker for your research.
Related products
Find out our relevant products for alternative pathway research.

C3d, Human, ELISA kit
Properdin, Human, ELISA kit
Complement factor D, Human, ELISA kit
C3c, Human, ELISA kit
C3, Mouse, mAb 11H9
C3b/iC3b/C3c, Mouse, mAb 2/11
Contact us for more information
At Hycult Biotech, we recognize the growing demand for advanced complement research tools that facilitate accurate analysis and targeted intervention in immune-related diseases. Our portfolio includes high-quality complement pathway inhibitors (how to inactivate complement), complement assay kits, and monoclonal antibodies designed to support drug discovery and translational research. On our website, you can search for products that match your research needs. Additionally, we are happy to provide expert advice. Feel free to contact us via the contact form.
Frequently asked questions
The AP is activated spontaneously through a mechanism known as “C3 tick-over.” In this process, native C3 undergoes continuous low-level hydrolysis, forming C3(H₂O), which can bind FB. Once bound, FB is cleaved by FD into Factor Bb and Ba, generating the fluid-phase C3 convertase C3(H₂O)Bb. This enzyme cleaves additional C3 into C3a and C3b. C3b can then bind to microbial or host cells, forming the surface-bound C3 convertase C3bBb. In addition, Properdin stabilizes this convertase, enhancing its activity.
The AP plays a dual role within the complement system both as an initiator and an amplifier of complement activation. It is continuously active at a low level via spontaneous hydrolysis of C3, allowing for immediate innate immune response in the absence of specific recognition molecules. Once triggered, the AP amplifies complement activation by forming the C3bBb convertase, which rapidly generates large amounts of C3b. Once C3b is generated, either via tick-over or through activation by the CP or LP, C3b can bind to target surfaces and initiate the formation of the AP-specific C3 convertase (C3bBb). This marks the transition to the amplification loop, in which the AP enhances complement activation by generating large amounts of additional C3b. Thereby, the AP not only initiates its own activation but also amplifies responses triggered by the CP and LP.
The AP differs from the classical and lectin pathways predominantly in how it is activated. While the classical pathway needs immune complexes and the lectin pathway depends on recognition of microbial carbohydrates, the AP activates spontaneously through a process called “C3 tick-over.” This involves continuous low-level hydrolysis of C3, leading to the formation of C3(H₂O), which can initiate complement activation without engagement of a recognition molecule.
Unlike the classical and lectin pathways, the AP is at a basal level active at a low level, serving as a constant surveillance mechanism of the innate immune system. This baseline activity allows the immune system to respond rapidly to pathogens or altered self-surfaces. The AP does not need recognition molecules, making it part of the innate immune system’s immediate defence.
Properdin functions as a positive regulator in the AP by stabilizing the C3 convertase complexes C3bBb and thereby enhancing the amplification of the complement response. Properdin binds to surface-bound C3bBb and extends the half-life of the convertase, supporting continued C3 activation. It also helps direct complement activation to pathogen surfaces by selectively binding to them, contributing to immune defence. By promoting targeted activation, complement deposition on host cells is prevented, thereby limiting unintended tissue damage.
FH is one of the most important regulators of the AP, essential for protecting host cells from unintended complement attack. It inhibits the AP at multiple levels: by accelerating the decay of the C3 convertase (C3bBb), by competing with FB for C3b binding, and by acting as a cofactor for FI to cleave C3b into inactive iC3b. These actions help prevent excessive complement activation in the fluid phase and on host surfaces.
In contrast, the FHRs lack regulatory functions like decay acceleration or cofactor activity but can compete with FH for binding sites on C3b and cellular surfaces. This competition can reduce FH-mediated control and promote complement activation, which is why FHRs are sometimes referred to as FH deregulators. Their function appears to support a more positive regulation of the AP, and they are implicated in modulating immune responses and contributing to complement-related diseases.
The AP contributes to disease pathogenesis by providing a constant state of low-level spontaneous activation, which, if not properly regulated, can lead to uncontrolled complement activation. This dysregulation plays a key role in many diseases not driven by autoantibodies, including inflammatory, autoimmune, and kidney disorders. Genetic variants in AP-related genes have been found in patients with these diseases, highlighting the pathway’s role in both health and pathology. The AP does not require a recognition molecule, making its activation dependent on the balance of local regulators and surface context.
Dysfunction of the alternative pathway is linked to several diseases, where its dysregulated activation drives or amplifies pathology. The following diseases are associated with the dysfunction of the alternative pathway:
- C3 glomerulopathy (C3G): Characterized by excessive deposition of C3 fragments in the glomeruli due to impaired regulation of the alternative pathway. This is often driven by mutations or autoantibodies that stabilize the C3 convertase or inhibit regulatory proteins like factor H. [3]
- Atypical hemolytic uremic syndrome (aHUS): Condition where defective control of the alternative pathway leads to complement-mediated injury of the microvascular endothelium, especially in the kidneys. Mutations in complement regulators (e.g., factor H, factor I, MCP) or presence of anti-factor H autoantibodies are often involved. [2][3]
- IgA nephropathy: In this common glomerulonephritis, secondary activation of the alternative pathway contributes to disease progression. Elevated FHR1 and FHR5 proteins promote C3 activation, enhancing glomerular injury. [3]
- Lupus nephritis: Although classically linked to the classical pathway, studies show that the alternative pathway amplifies complement activation and tissue damage in lupus, particularly in mouse models deficient in factor B or D, which show protection from renal injury. [1][2]
- Cardiovascular disease (CVD): In patients with chronic kidney disease (CKD), increased alternative pathway activation markers like factor D and Factor Ba correlate with endothelial dysfunction and elevated CVD risk. This suggests the pathway contributes to vascular inflammation and atherosclerosis. [3]
- Age-related macular degeneration (AMD): Strongly associated with polymorphisms in the CFH gene, which impair regulation of the alternative pathway at the retinal pigment epithelium, leading to complement-mediated retinal damage. [2][3]
[1] Satyam A, Hisada R, Bhargava R, Tsokos MG, Tsokos GC. Intertwined pathways of complement activation command the pathogenesis of lupus nephritis. Transl Res. 2022 Jul;245:18-29. doi: 10.1016/j.trsl.2022.03.005. Epub 2022 Mar 14. PMID: 35296451; PMCID: PMC9167748.
[2] Brandwijk RJMGE, Michels MAHM, van Rossum M, de Nooijer AH, Nilsson PH, de Bruin WCC, Toonen EJM. Pitfalls in complement analysis: A systematic literature review of assessing complement activation. Front Immunol. 2022 Oct 18;13:1007102. doi: 10.3389/fimmu.2022.1007102. PMID: 36330514; PMCID: PMC9623276.
[3] Jalal DI, Thurman JM, Smith RJ. Chronic kidney disease enhances alternative pathway activity: a new paradigm. J Clin Invest. 2025 May 1;135(9):e188353. doi: 10.1172/JCI188353. PMID: 40309771; PMCID: PMC12043098.